Keywords: Matching · Structural Equation Modeling · Decision Trees · Machine Learning
Understanding the causal effect of a treatment has historically been of great scientific interest and remains one of the most frequently pursued objectives in scientific research today. The gold standard for evaluating treatment effects is the randomized controlled trial, where the researcher randomly assigns treatment status to each individual. The benefit of this approach is that the causal effect of the treatment can be estimated by simply comparing outcomes between those who were treated and those who were not (Greenland, Pearl, & Robins, 1999). Random assignment of treatment guarantees that, on average, the treated and untreated individuals will be equal on all potential confounding variables, both measured and unmeasured. Eliminating the possibility of confounding clears the way for a direct comparison to be made.
However, random assignment is not always possible. This can be for ethical reasons, since researchers cannot, for example, force participants to smoke to investigate the effects of smoking. It can also be for practical reasons, where the researcher cannot control the assignment of a treatment. For example, researchers cannot randomly assign depression to some participants, enact a law or policy in a randomly assigned jurisdiction, or choose where their participants live. An observational study, where treatment is not randomly assigned, may be the only available option in these cases. Unlike randomized controlled trials, direct comparisons between treated and untreated individuals in an observational study cannot be made as easily. This is because treated and untreated participants may not be equal in all other characteristics, creating the potential for confounding effects. In fact, it may be differences in these very characteristics that lead some participants to select treatment, making the estimation of the treatment’s effect less straightforward. To estimate a treatment’s effect, it must first be defined, which we do in the context of the potential outcomes framework.
The foundations for the potential outcomes framework were laid out by Neyman, Iwaszkiewicz, and Kolodziejczyk (1935) and further developed by Rubin (1974), resulting in it also being called the Rubin Causal Model, Neyman-Rubin Causal Model, and Neyman-Rubin counterfactual framework of causality. The model can be conceptualized as follows. Let $Y_{1i}$ be the potential outcome of individual $i$ if they received the treatment and $Y_{0i}$ be the potential outcome of individual $i$ if they did not receive the treatment. The observed score $Y_i$, can be written as
|
| (1) |
where $W_i = 1$ if the individual received treatment and $W_i = 0$ if they did not. $W_i$ simply acts as an indicator variable denoting the receipt of treatment. The term treatment here and throughout the paper is used rather loosely and can be used interchangeably with exposure.
The effect of the treatment is simply $Y_{1i} - Y_{0i}$, the difference between the potential outcomes if the individual had received treatment and if they had not. The fundamental problem of causal inference, as stated by Holland (1986), is that it is impossible to observe both $Y_{1i}$ and $Y_{0i}$ for the same individual. If the individual received treatment, we can observe $Y_{1i}$, but not its counterfactual, $Y_{0i}$ . The inverse is also true: if the individual did not receive treatment, we can observe $Y_{0i}$ , but not its counterfactual, $Y_{1i}$. Therefore, it is impossible to observe the effect of the treatment on the individual. As an example, we can see that it is impossible to observe the effect of divorce on a child’s academic test scores because at a given moment in time, the parents can either be divorced or not divorced, but not both. We cannot observe the test scores under both conditions, so we cannot observe the effect of divorce on that child’s scores.
Though we cannot observe the effect of the treatment on a given individual, we can estimate the average treatment effect (ATE) on a population. The ATE is the average effect expected from taking a population where no individuals received the treatment and providing the treatment to all of them (Austin, 2011). The ATE is defined as $ATE= E(Y_{1i}-Y_{0i})=E(Y_{1i})-E(Y_{0i})$, where $E(\cdot )$ is the expected value operator. Conceptually, this implies that although we cannot observe the treatment effect at the individual level, we can do so at a population level by using the average of the untreated participants as a proxy for the unobservable counterfactual (Guo & Fraser, 2010). A related effect of interest in this paper is the conditional average treatment effect , or CATE, (Abrevaya, Hsu, & Lieli, 2015), defined as $CATE= E(Y_{1i}-Y_{0i} \vert \boldsymbol {X}_i)$, where $\boldsymbol {X}_i$ is a vector of covariates. The CATE allows us to evaluate heterogeneity in treatment effects between subpopulations, for example, allowing for separate estimation of the ATE in males and females if they are believed to be different.
One important assumption of the potential outcomes framework is the Stable Unit
Treatment Value Assumption, or SUTVA (Rubin, 1980, 1986). It represents
the assumption that the potential outcomes would be the same no matter
how an individual came to be assigned to a treatment, and no matter what
treatments are received by other individuals. It assumes that neither treatment
assignment mechanisms nor social interactions affect potential outcomes. Another
assumption, one we give more attention due to the focus of this paper, is
known as the strong ignorability assumption (Rosenbaum & Rubin, 1983).
Treatment assignment is strongly ignorable if two conditions collectively hold. The
first condition is $(Y_0,Y_1) \perp W \vert \boldsymbol {X}$, that treatment assignment is independent of the potential
outcomes conditional on covariates. The second condition is $0 The necessity of the conditional independence piece of the strong ignorability
assumption becomes evident when considering the necessary conditions for using
untreated participants as a proxy for the counterfactual. To estimate the
ATE by taking the difference between the averages of treated and untreated
participants, we implicitly assume that the average scores produced by the
untreated participants are an unbiased estimate of what the average scores
produced by the treated participants would have been had they not received
the treatment. In doing so, we must ensure that the treated and untreated
participants are similar in relevant characteristics, so that the untreated
participants can serve as a faithful representation for their treated counterparts. For
example, if the treated group contained only males and the untreated group
contained only females, using the untreated group as a proxy for the treated
group might not produce a fair comparison, depending on what is being
studied. This is why randomized controlled trials are considered the gold
standard: random assignment ensures that, on average, all such possible
confounders are balanced, making the treated and untreated participants
comparable.
As pointed out by Thoemmes and Kim (2011), the strong ignorability
assumption cannot be empirically tested. This is because treatment assignment must
be conditionally independent of all relevant covariates both observed and unobserved,
and it is not possible to empirically verify that variables that are not collected do not
play a role. As such, researchers who attempt to justify this assumption are limited to
making a convincing argument that they have measured the relevant covariates and
showing that these are balanced across treated and untreated participants. The
most common way of demonstrating balance in an observed covariate across
groups is via a standardized mean difference. This takes the form of the mean
difference in the covariate between groups (in absolute value) divided by either a
pooled standard deviation or an unpooled standard deviation of one of the
groups.
A standardized mean difference of 0 would indicate the covariate has the same
mean across groups. However, there is no universally agreed upon metric for judging
how small a nonzero standardized mean difference must be to be considered
negligible enough for the groups to be considered balanced on the covariate
for practical purposes. Many recommendations exist in the methodological
literature. Harder, Stuart, and Anthony (2010) use a value less than 0.25,
based on a suggestion by Ho, Imai, King, and Stuart (2007). Austin (2011)
suggests a stricter value of less than 0.1, based on work by Normand et
al. (2001). Leite, Stapleton, and Bettini (2018) point out that for educational
research, the What Works Clearinghouse Procedures and Standards Handbook
(version 4.0) requires a value less than 0.05 without additional covariate
adjustment, or between 0.05 and 0.25 with additional regression adjustment (U.S.
Department of Education, Institute of Education Sciences, & What Works
Clearinghouse, 2017).
Analyzing standardized mean differences is reasonable when attempting to balance
across demographic covariates such as sex, age, race, etc. Yet some characteristics do
not lend themselves well to being assessed in this way. Consider an example where we
are interested in evaluating the effects of a breakup from a romantic relationship (the
treatment) on life satisfaction (the outcome). For simplicity, let us assume that we
only collect data from one partner per couple. Putting demographics aside,
affect might be an important covariate to balance on. However, ensuring that
couples who do and do not break up have the same average affect might not
be especially useful. Stability of affect has been shown to be predictive of
whether couples remain together or break up (Ferrer, 2016; Ferrer, Steele, &
Hsieh, 2012). That is, fluctuations in affect are what need to be balanced, not
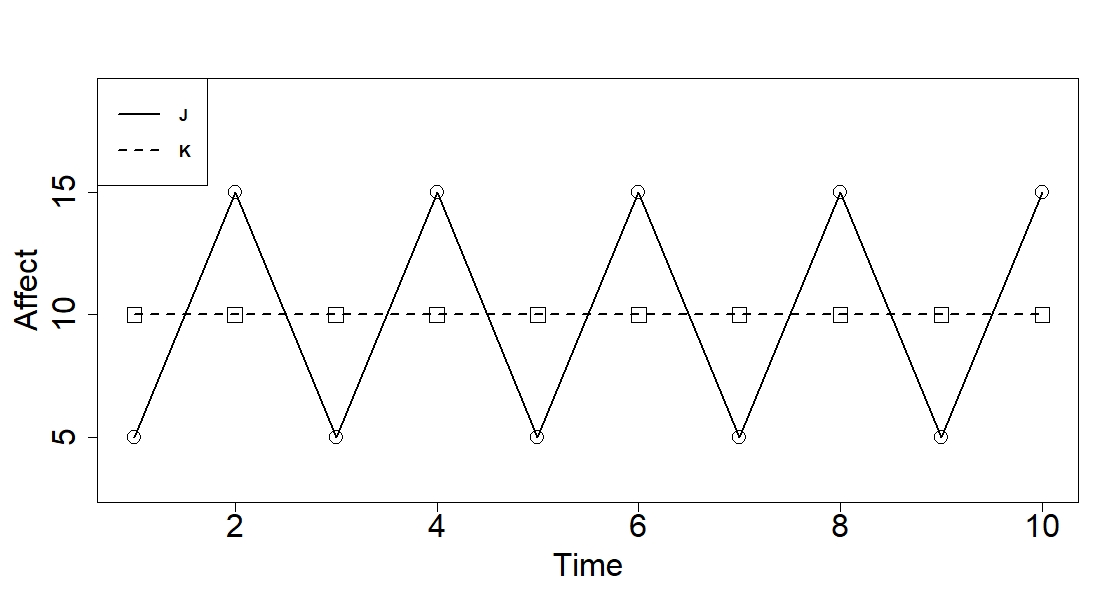
simply average affect. Consider the plot given in Figure 1 of two hypothetical
individuals, J and K, and their affect over time. J has highly variable affect,
whereas K has relatively stable affect. Based on the aforementioned research,
J is more likely to experience a breakup, given their instability. However,
both J and K have the same average affect. Imagine a treatment group filled
with individuals like J and an untreated group filled with individuals like K.
According to the standardized mean difference, these two groups would be
balanced across affect, because they have the same mean affect. The fact that
they have different patterns with regard to the variability would be entirely
missed.
The literature does recommend that covariates should be balanced across groups
on not just the mean, but the distribution of the variables (Austin, 2011; Ho et
al., 2007). Researchers are encouraged to examine higher-order moments, as well as
interactions between covariates. Graphical methods are often used to make these
comparisons, including quantile-quantile plots, boxplots, density plots, etc. Though
visualizations can be helpful for univariate or even bivariate data, they become less
useful with higher-dimensional data, as in our example. Furthermore, in this case,
they do not quite address the issue directly. We would like to balance on stability of
affect, which is not entirely captured by either univariate higher order moments or
interactions.
Although conventional approaches can be useful when balancing on demographic
variables and other such covariates, they are not as well suited for balancing on more
complex functions of the data, such as stability of affect. This paper seeks to develop
an approach that allows us to balance on more flexibly defined characteristics of
interest. We begin by reviewing some classic and recent approaches to matching. We
then provide an introduction to structural equation model trees and their variations.
Drawing from these, we propose our own algorithm, Causal Mplus Trees, and
describe its implementation. We then conduct two small simulation studies
demonstrating our algorithm’s effectiveness and an empirical analysis of COVID-19
data. We conclude with a discussion of practical recommendations and future
directions.
Thus far we have discussed ways to evaluate whether treated and untreated
participants are balanced on covariates. If they are found to be unbalanced, we can
turn to statistical approaches to balance them. A natural initial thought would be to
use ordinary least squares regression, conditioning on covariates within the model.
However, Berk (2004) points out that simply calculating a conditional distribution of
the outcome is not sufficient to draw causal inference and that stronger assumptions
are needed.
A popular alternative is to use propensity scores, defined as the probability of
treatment conditional on observed covariates (Rosenbaum & Rubin, 1983). It has
been shown that propensity scores can balance treated and untreated participants in
the sample, and that both treatment assignment as well as observed covariates are
conditionally independent given the propensity score (Rosenbaum & Rubin, 1983).
This implies that for participants with the same propensity score, the mean difference
in the outcome between treated and untreated participants is an unbiased estimate of
the ATE at that propensity score (Guo & Fraser, 2010). Propensity scores are
typically calculated using logistic regression, with the observed covariates
predicting treatment status ($W$). The estimated regression coefficients are then
used as weights in a model predicting the probability of treatment for each
individual. The estimated propensity score is this predicted probability of
treatment.
Once propensity scores have been calculated, they can be used in various ways,
including propensity score matching (Rosenbaum & Rubin, 1985), stratification on
the propensity score (Rosenbaum & Rubin, 1984), and inverse probability of
treatment weighting using the propensity score (Hirano & Imbens, 2001). Of these
three, propensity score matching seems to eliminate more of the systematic differences
in covariates (Austin, 2009) and also seems to be the most popular (Thoemmes &
Kim, 2011), so we limit our focus to propensity score matching. Propensity score
matching involves finding treated and untreated participants with similar propensity
scores to use as each other’s counterfactuals. According to Austin (2011)
and the systematic review conducted by Thoemmes and Kim (2011), the
most commonly used form of matching is 1:1 matching, where each treated
participant is matched with a single untreated participant, forming a pair.
Thoemmes and Kim (2011) found that the most popular way to do this in the social
sciences was to use greedy matching, in which a treated subject is selected
at random and the untreated subject with the closest propensity score is
paired with them. The process is repeated until all treated subjects have a
match. This is in contrast to optimal matching, where matches are selected
to optimize the distance between propensity scores for the entire sample,
which has been shown to perform comparably to greedy matching (Gu &
Rosenbaum, 1993). The 1:1 matching scheme produces pairs of treated and
untreated participants who should in theory be balanced on the propensity
scores. The ATE can then be estimated simply by performing a paired $t$ test
(Austin, 2011).
Of course, one must still ensure that the propensity scores are balanced across
treated and untreated participants. If they are not, it is recommended that
the logistic regression model be iteratively refined by including nonlinear
terms and interactions between covariates until balance has been achieved
(Austin, 2011; Rosenbaum & Rubin, 1984, 1985; West et al., 2014). Latent
variable models can be used to calculate propensity scores by balancing on latent
covariates whose scores are estimated via factor score estimation (Raykov, 2012), or
by using structural equation modeling to estimate propensity scores directly (Leite et
al., 2018). Machine learning techniques including bagging, boosting, trees, and
random forests, have also been used for the estimation of propensity scores (Lee,
Lessler, & Stuart, 2010).
A recent alternative to propensity score matching is the causal tree approach
proposed by Athey and Imbens (2016). Essentially, they use decision trees to
partition the sample into groups of individuals who are similar on important
dimensions. They then treat these groupings as matched, and use them to estimate
the ATE. Decision trees (Breiman, Friedman, Olshen, & Stone, 1984), use recursive
partitioning to separate a predictor space into regions that are as homogeneous as
possible on a target variable of interest. Binary splits are made on predictors (e.g.
female vs. male, age $\leq $ 60 vs. age $>$ 60, etc.), splitting the sample into two nodes. All
possible splits are made on all predictors, and the split that makes the resulting
samples in each node as homogeneous as possible is presented as a candidate
split. If this split exceeds a predetermined fit criterion, the split is made,
partitioning the sample into the two daughter nodes. Otherwise, the split
is not made, and the parent node becomes a terminal node. The process
continues recursively on each daughter node until all nodes are terminal nodes.
We refer readers to Serang et al. (2021) for additional description of the
procedure.
Decision trees are most often used for prediction of a target variable. The critical
insight of Athey and Imbens (2016) is that trees have a natural proclivity
for creating homogeneous subgroups. Instead of trying to predict a target
variable, we can substitute the vector of covariates, $\boldsymbol {X}$. The tree will then produce
terminal nodes where the observations in each terminal node are as similar
as possible on the covariates, achieving the same aim as matching. Each
terminal node is characterized by splits on predictors (separate from $\boldsymbol {X}$) that
define membership in that node. In what they call an honest approach to
estimation, the authors recommend that these subgroup definitions be applied
to a fresh holdout sample not involved in the construction of the tree, to
create subgroups using the new data. CATEs (ATEs conditional on subgroup
membership) can then be estimated in each subgroup via mean differences between
treated and untreated participants within the subgroup. Causal inference can
also be drawn using standard approaches, such as an independent-samples $t$
test.
The advantage of causal trees over propensity score methods is that one
need not worry about the estimation of or balancing on propensity scores.
Propensity scores only serve as a middleman in propensity score matching,
and causal trees use the properties of decision trees to bypass them entirely.
Additionally, causal trees easily accommodate heterogeneity in causal effects.
In our running example, we wish to match on stability of affect. If we use
demographic variables as splitting variables in the tree, we can potentially find
subgroups defined by these demographic characteristics (e.g. sex, age, etc.)
that have different levels of affect stability. The causal tree approach would
then allow us to estimate the causal effect of a breakup separately in each of
these subgroups, as well as compare them to see if the causal effect differs by
subgroup.
One limitation of causal trees as described is that they assume we wish to match on
observed covariates. However, stability in our example is not an observed variable in
the data: it is a characterization based on a pattern. One way to characterize stability
for the data in our example would be to fit a simple intercept-only growth curve
model and examine the residual variance. A model fit to individuals such as J would
produce a large residual variance, whereas a model fit to individuals like K
would yield a relatively small residual variance. Thus, stability of a group can
be characterized by model-based parameter estimates, in lieu of observed
variables.
To do this within the causal tree framework, we would need a mechanism to fit a
model within each node. For longitudinal models, we can use an approach like the
nonlinear longitudinal recursive partitioning algorithm proposed by Stegmann,
Jacobucci, Serang, and Grimm (2018), which allows the user to fit linear and
nonlinear longitudinal models within each node. A more general approach is the
structural equation model tree (SEM Tree) proposed by Brandmaier, Oertzen,
McArdle, and Lindenberger (2013), which allows for structural equation models
(SEMs) to be fit within each node. A benefit of the latter is the flexibility of the
SEM framework, which can accommodate a wide range of models, including
many longitudinal models, via latent growth curve modeling (Meredith &
Tisak, 1990).
The logic of SEM Trees is similar to that of standard decision trees, with some
minor variations. A prespecified SEM is first fit to the full sample, and the minus two
log-likelihood ($-2LogL$) is calculated. Then, the $-2LogL$ for the candidate split is calculated. Since
the split can be conceptualized as a multiple group model (Jöreskog, 1971), the $-2LogL$ for
the split is simply the sum of the $-2LogL$ values for each daughter node. A likelihood ratio
test is then conducted with these two $-2LogL$ values. If it rejects, the split is made. As in
other decision trees, this process is recursively repeated until all daughter nodes are
terminal nodes. Unlike conventional decision trees, terminal nodes in SEM Trees do
not provide a predicted proportion or mean. Rather, each terminal node is
characterized by a set of parameter estimates for the SEM fit to the sample
in that node. In this way, SEM Trees can be used to identify subgroups of
people who are similar in that they can be represented by a set of parameter
estimates that is distinct from the parameter estimates that characterize those
in other nodes. SEM Trees can therefore identify subgroups with distinct
patterns of stability, growth, or other patterns reflected in the parameter
estimates.
The SEM Trees algorithm is implemented in the semtree (Brandmaier, Prindle, &
Arnold, 2021) package in R (R Core Team, 2020). The SEMs are fit in either the
OpenMx package (Neale et al., 2016) or the lavaan package (Rosseel, 2012). The
OpenMx package is flexible but challenging to use, especially for casual users, given the
need to specify the entirety of the model with limited defaults. The lavaan package is
much easier to use given the ease with which one can specify models, however it is
currently more limited in the scope of the models it can fit. The MplusTrees package
(Serang et al., 2021) is an implementation of SEM Trees which uses Mplus
(Muthén & Muthén, 1998-2017) to fit the models, the rpart package (Therneau &
Atkinson, 2018) to perform the recursive partitioning needed to grow the trees, and
the MplusAutomation package (Hallquist & Wiley, 2018) to interface between R
and Mplus. MplusTrees capitalizes on the wide variety of complex models
that can be specified in Mplus, the ease with which they can be specified,
and the currently superior estimation algorithms it uses for fitting these
models.
The Mplus Trees algorithm itself (Serang et al., 2021) is very similar to the SEM
Trees algorithm (Brandmaier et al., 2013). However, one key difference is the
criterion used for splitting. Although the MplusTrees package also has the capability
to split using the likelihood ratio test, this is not the primary method. Instead, Mplus
Trees uses a complexity parameter, cp. This cp parameter is a proportion
specified in advance by the user. A split will be made if that split improves on
the -2LogL of the full sample (the parent node) by at least cp times that
-2LogL. Smaller values of cp result in more splits since a relatively smaller
improvement in the -2LogL is needed for a split to be made, whereas larger values
lead to fewer splits. As such, the use of cp serves more as a heuristic than a
formal test based on statistical significance. Ideally, cp would be selected
by cross-validation, and this functionality is available in the MplusTrees
package. However, long computational times may require users to simply
try a handful of cp values and select the most appropriate one given the
context.
We now propose our own matching algorithm, Causal Mplus Trees, using Mplus Trees
to create causal trees that match on parameters from an SEM, and estimating CATEs
in a holdout sample. We begin by first randomly partitioning the dataset into two
parts, one subsample to perform the matching and the other to perform the
estimation of the CATEs. In most cases, the matching subsample will require more
participants, since fitting an SEM and building a decision tree is more sample
intensive than estimating a mean difference. We suggest devoting 80% of the sample
to the matching subsample and 20% to the estimation subsample, though this ratio
can be adjusted depending on the complexity of the SEM, the overall sample size,
etc.
Beginning with the matching subsample, we can partition $\boldsymbol {X}$ into two parts: $\boldsymbol {X}_M$, the
modeled covariates modeled in the SEM whose parameters we wish to match on, and $\boldsymbol {X}_S$,
the splitting covariates we want to split on in the recursive partitioning process which
define the subgroups of the tree’s terminal nodes. Guidance for whether a covariate
should be a modeled covariate or a splitting covariate is provided in the
discussion. Let $M$ be an SEM with parameters $\boldsymbol {\theta }$ that produces $\boldsymbol {X}_M$, so that $M(\boldsymbol {\theta }) = \boldsymbol {X}_M$. In our
running example, $M$ would be the intercept-only growth model and $\boldsymbol {\theta }$ would be its
parameters. For properly specified $M$, $\boldsymbol {X}_M$ can be used to estimate $\boldsymbol {\theta }$, resulting in
parameter estimates $\hat {\boldsymbol {\theta }}$. Using Mplus Trees, we can build a tree that matches on $\hat {\boldsymbol {\theta }}$,
with groups (terminal nodes) defined by their covariate patterns on $\boldsymbol {X}_S$. The
treatment assignment information, $W$, is not provided to the recursive partitioning
algorithm and so the tree is built blind to $W$. In the estimation subsample,
we can divide participants into groups according to the splits found by the
tree. Within each group, we can estimate the CATE as defined before by
taking the difference between the means of the outcomes of the treated and
untreated participants in each group. Since we are using a fresh sample, we
can draw inference using hypothesis tests such as an independent-samples $t$
test or another suitable alternative. We can also test whether the CATE
differs by group by testing the interaction effect in a two-way independent
ANOVA.
As a proof of concept for Causal Mplus Trees, we performed two small simulation
studies. The simulation studies were conducted in R using the lavaan package to
simulate data and the MplusTrees package for analysis. Readers are referred to the
package documentation for details regarding the implementation of the algorithm in
the software. Each simulation consisted of 1,000 replications.
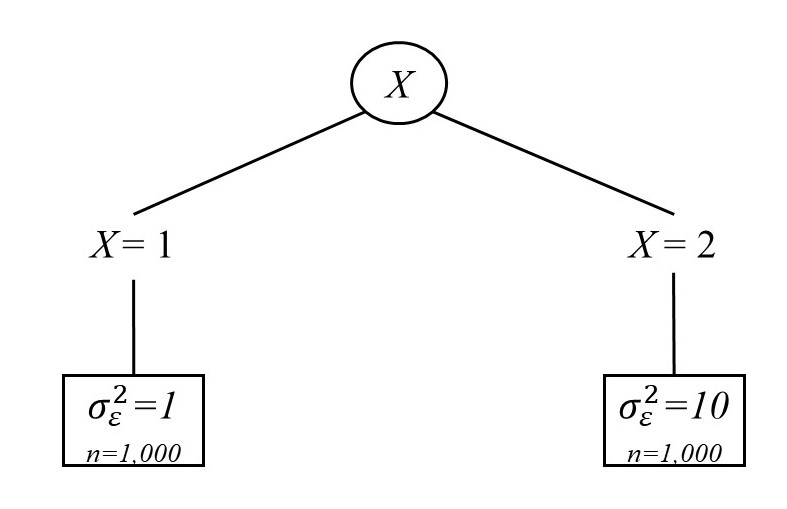
The first simulation mapped onto our running example regarding stability
of affect. Each sample consisted of N = 2,000 individuals, 1,000 in each of
two groups. The data were generated from an intercept-only (no growth)
model with 10 time points. The intercept had a mean of 10 with a variance of
1. The only difference between the groups was in the residual variance, $\sigma _\epsilon ^2$.
One group had a residual variance of 1 (the group with stable affect), and
the other had a residual variance of 10 (the group with unstable affect).
The group memberships were identified by a dichotomous covariate, used
as a splitting variable. Thus, the tree matched on the growth curve, using
the group membership to split. Within each group, treated and untreated
participants were evenly split (500 each). A diagram of this population tree is given
in Figure 2. For the stable affect group, outcomes were generated using a
standard normal distribution, $N(0,1)$, for the untreated group and a $N(0.5,1)$ distribution for
the treated group, to represent a medium-sized CATE. However, for the
unstable affect group, the outcome distributions were flipped, with the untreated
group’s outcome being generated from a $N(0.5,1)$ distribution, whereas the treated
group’s outcome was generated from a $N(0,1)$ distribution. In this way, although the
ATE for the full sample was 0, the CATE for each group was 0.5 in absolute
value.
It should be noted that these groups are, from the start, balanced on the modeled
covariates. Since the growth curve variables were all generated to have a mean of 10,
they would be considered balanced according to the standardized mean difference.
Thus, if one were to follow conventional procedure, propensity scores would not be
needed here, and the estimation of the ATE would consist of simply the mean
difference between treated and untreated participants, which would be 0 on
average.
The Causal Mplus Trees algorithm was implemented as described in the prior
section, with 80% of the sample (1,600 individuals) used for matching and 20% (400
individuals) used to estimate CATEs. A $cp$ value of .01 was used to split, with a
minimum of 100 individuals required to consider splitting on a node. Each terminal
node was also required to have at least 100 individuals within it. For each replication,
the CATE was estimated in each group using an independent samples $t$ test. A
two-way independent ANOVA was also conducted to determine if CATEs differed by
group.
Overall, the results demonstrated the effectiveness of the algorithm. Across all
replications, 94.5% of CATEs were detected. Additionally, 99.8% of the interactions
from the two-way ANOVA were detected, showing that the algorithm can detect
differences in CATEs by group. As a comparison, we also analyzed these data as
they would have been analyzed using the conventional approach. Since the
covariates were on average balanced according the standardized mean difference,
the ATE would have been estimated by using the full sample to estimate
the mean difference between treated and untreated participants. Despite a
sample size of 2,000 to do this (relative to the only 400 available to Causal
Mplus Trees after performing the matching), only 3.4% of datasets yielded
statistically significant ATEs, consistent with a nominal false positive rate of
5%.
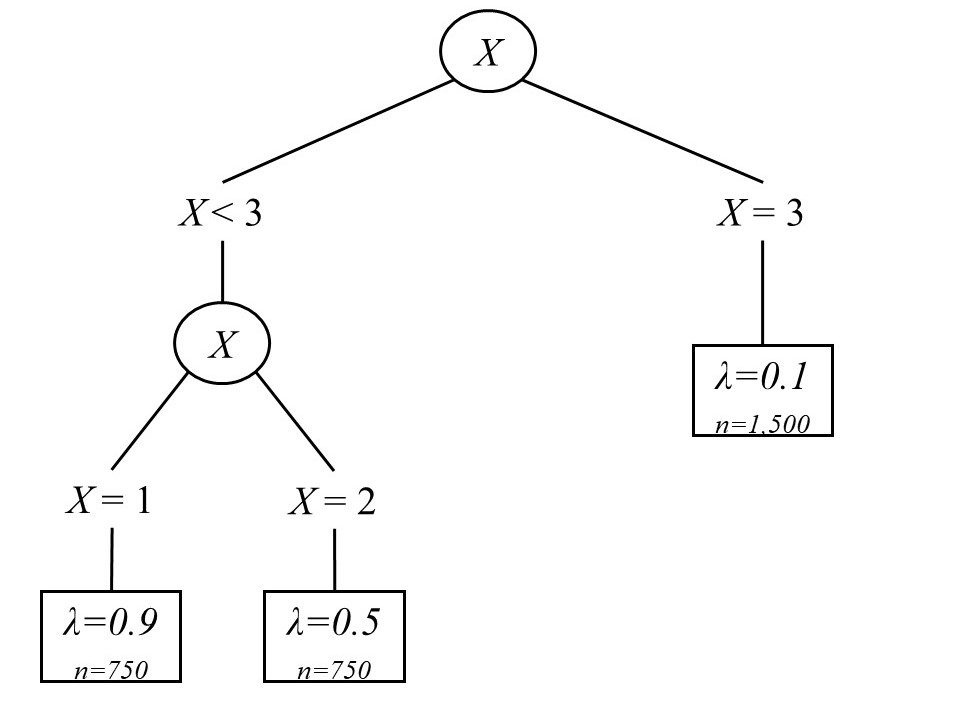
The second simulation study is similar to the first, but used a measurement model as
opposed to a longitudinal model for the matching. For the second study,
each sample consisted of N = 3,000 individuals, divided into three groups.
One group (the small loading group) contained 1,500 individuals, while the
remaining two groups (the medium and large loading group) each contained 750.
Data were generated from a one-factor confirmatory factor analysis model
with 15 items. Factor variances were fixed to 1, and uniquenesses were also
simulated to be 1. As implied above, the only differences were in the loadings,
$\lambda $. In the small loading group, all loadings were simulated to be 0.1, in the
medium loading group they were 0.5, and in the large loading group they
were 0.9. The model was generated to reflect the case where items are more
related to a latent construct for some people than for others. If the latent
variable were a psychological disorder, this would map onto the idea that the
items better reflect the presence of that disorder in some groups relative to
others.
As with the previous simulation study, a single splitting covariate denoting group
membership was used as the splitting variable, albeit with three values given the three
groups. Figure 3 shows a diagram for this population tree. As with the other
simulation study, each group was evenly divided on treated and untreated
participants. In the small loading group untreated participants had outcomes
generated from a $N(0,1)$ distribution, whereas the treated group’s outcome was generated
from a $N(0.5,1)$ distribution. In the medium and large loading groups this was reversed:
untreated participants had outcomes from a $N(0.5,1)$ distribution whereas treated participants
had outcomes from a $N(0,1)$ distribution. In this way, these samples too had an average
ATE of 0, in addition to being on average balanced on the modeled covariates
according to the standardized mean difference, since all items had an average score of
0.
The algorithm again used 80% of each sample (2,400 participants) for matching
and 20% (600 participants) for estimation. As before, a minimum of 100
individuals was required to consider splitting a node and in each terminal node,
however this study used a cp value of .001. Unlike the previous study where the
split was made in every replication, the algorithm had some slight trouble
finding all the groups in this study. All three groups were found in 92.7% of
simulations, but only two groups were found in the remaining 7.3%. Among all the
groups found, 88.3% of the CATEs were detected, along with 99.7% of the
interactions. Alternatively, when using the entire sample to calculate the
ATE, only 3.5% of simulations yielded significant results. These results are
similar to those found in the first simulation study. Taken together, they
show that the Causal Mplus Trees algorithm is able to estimate CATEs and
support hypothesis testing to determine their statistical significance. It can
also determine whether the CATEs differ by group. Notably, CATEs were
found in the absence of ATEs, with modeled covariates already balanced
across treated and untreated participants according to the standardized mean
difference.
As an illustration of how Causal Mplus Trees can be used in practice, we present an
analysis of COVID-19 data. The dataset contains information from four
different sources: public health data from the COVID-19 Data Repository by
the Center for Systems Science and Engineering (CSSE) at Johns Hopkins
University (Dong, Du, & Gardner, 2020), demographic data from the 2010 US
Decennial Census (U.S. Census Bureau, 2010), governor’s party information
obtained from the National Governors Association Roster (National Governors
Association, 2020), and mobility data from Unacast, a location data analytics
company (Unacast, 2020).
To capture how individuals’ travel activity patterns responded to the spread of
COVID-19, we utilized Unacast’s measure of the change in average distance traveled.
Travel distance was measured using the GPS positions of millions of mobile devices
and aggregated each day to a county-level average. For a detailed overview of
variable construction and discussion of potential sources of bias, see Sears,
Villas-Boas, Villas-Boas, and Villas-Boas (2020). The data were analyzed
at the county level, consisting of 3,030 counties or county-equivalents from
all 50 US states except Alaska. This represents over 95% of counties in the
US.
The goal of this analysis was to estimate the CATE of the governor’s party
(Democrat or Republican) on mobility in counties matched on the trajectory of
COVID-19 cases early in the pandemic. We sought to answer the question: “for
counties with similar trajectories of the rise in COVID-19 cases from March through
June 2020, could differences in mobility in July 2020 be attributed to the governor’s
party?” Prior studies reveal strong links between political partisanship and the
adoption of stay-at-home and social distancing orders as well as changes in
residents’ travel behavior and time spent at home (Adolph, Amano, Bang-Jensen,
Fullman, & Wilkerson, 2020; Allcott et al., 2020; Brzezinski, Deiana, Kecht, &
Van Dijcke, 2020; Gadarian, Goodman, & Pepinsky, 2020). We provide
a complementary analysis allowing us to understand whether the effect of
gubernatorial political alignment extended beyond stay-at-home adoption
timing to continued behavioral changes among constituents. Our analysis also
examined how this effect differed across counties depending on demographic
characteristics.
Case trajectories were modeled using the cumulative cases in the county divided
by the population per 10,000 residents, hereafter referred to as COVID rates. COVID
rates were calculated weekly from March 9, 2020 (around when states began reporting
their first cases) until June 29, 2020, resulting in 17 time points of data per county.
The SEM fit within each node of the tree was the logistic growth model given
by
where $COVID_i$ is the COVID rate for county $i$, $\beta _{1i}$ is the county-specific COVID rate
when the “curve has flattened” (the upper asymptote), $t$ is the number of
weeks ($t =$ 1, 2, …, 17), $\gamma $ is the inflection point, $\alpha $ is the rate of change, and $\epsilon $ is the
residual. The model was specified using Taylor-series approximation (Browne &
Toit, 1991; Grimm & Ram, 2009) with equal residual variances across time, $\sigma _\epsilon ^2$, to aid
estimation.
We used six demographic splitting variables: population (the total population of
the county), white (the percentage of non-Hispanic Whites), age65_older (the
percentage of people ages 65 years and older), median_inc (the median household
income), bachelors (the percentage of people with at least a bachelor’s degree), and
rural (the percentage of the population considered rural). To reduce the
computational burden of the algorithm, we reassigned values from 1 to 4 to each of
these splitting covariates depending on the quartile in which they fell relative to the
other counties.
In implementing the Causal Mplus Trees algorithm, we used 2,424 counties to
match the data and the remaining 606 to estimate the CATEs. We required that a
minimum sample size of 300 was required to both attempt a split and to remain in
each terminal node. A cp value of .01 was used to split. The tree grown from the
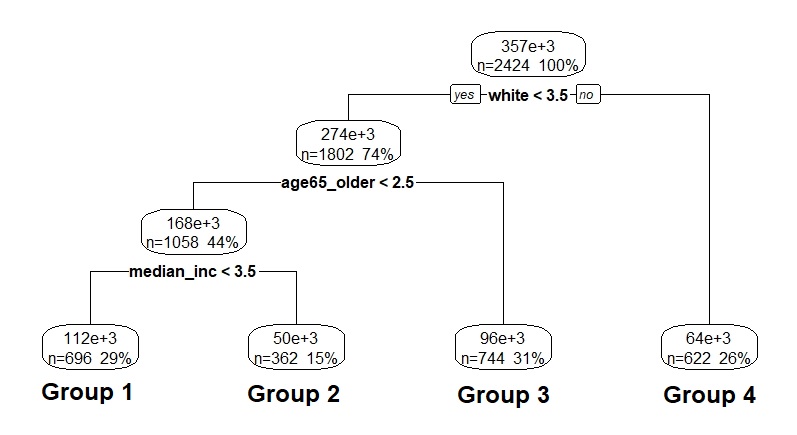
training data is given in Figure 4, with corresponding parameter estimates provided
in Table 1. Group 1 consisted of those in the bottom three quartiles ($<$93.1%) on white,
below the median ($<$17.2%) on age65_older, and in the bottom three quartiles of
median_inc ($<$$53,601). It contained 29% of the counties, and was characterized by the
highest asymptote, 61.33 cases per 10,000. Group 2 was made up of those in the
bottom three quartiles ($<$93.1%) on white, below the median ($<$17.2%) on age65_older,
but in the top quartile of median_inc ($>$$53,601). It represented 15% of the
counties, and was characterized by the second highest asymptote, 50.19 cases per
10,000. Group 3 contained those in the bottom three quartiles ($<$93.1%) on
white, but above the median ($>$17.2%) on age65_older. This group had 31% of
counties, with the second lowest asymptote, 35.88 cases per 10,000. Group 4
consisted of those in the top quartile ($>$93.1%) on white, with 26% of counties
and the lowest asymptote at 19.04 cases per 10,000. Group 4 also happened
to be the most rural and least populated, potentially explaining the low
asymptote.
Governor’s party (with Republican arbitrarily selected as the treatment) was used
as the treatment variable in part because much of the policy, coordination, and
messaging thus far has occurred via executive action at the state level. The outcome,
mobility, was operationalized as the change in average distance traveled, or
CADT. CADT for each day in July was calculated as the county-day level
percentage point change in average travel distance relative to that day-of-week’s
average in early 2020 (average for Feb 10 to March 8, prior to the presence of
COVID-19 in the US). Accordingly, a value of –3 indicates a 3 percentage point
decline in average travel distance relative to baseline levels. A positive value of
CADT signals that residents of that county increased their travel distances
relative to their pre-COVID-19 patterns, whereas a negative value indicates
reduced travel distances (that can occur through reductions in both the
distances traveled per trip as well as the overall number of trips taken). Each
county’s average CADT for July was estimated by taking the mean of the daily
CADT for each day from July 1, 2020 until July 31, 2020. The estimate of
the CATE in each group, along with corresponding information, is given in
Table 2.
Of the four groups, the only one with a statistically significant CATE was
Group 2, where counties in states with Democratic governors had an average
CADT that was 6.46 percentage points less than counties in states with
Republican governors $t$(108.76) = -2.84, $p$ = .006. Group 2 was on average the most
populous, least rural group of the four, as well as the most educated with
highest median incomes. As such, Group 2 contained the country’s more
metropolitan areas. We interpret this result to mean that in metropolitan counties
matched for COVID rates, people in counties in states with Democratic
governors traveled 6.5 percentage points less in July than people in comparable
counties in states with Republican governors. Of note, the two-way independent
ANOVA found that in the estimation subsample, a significant main effect of
party was not found $F$(1, 598) = 3.76, $p$ = .053, whereas a main effect of Group
$F$(3, 598) = 13.45, $p <$ .001, and an interaction $F$(3, 598) = 3.41, $p$ = .017 were.
This suggests that the party effect is more prominent for more metropolitan
counties, but would be obscured if examining the country as a whole. The
mean difference between parties in CADT for all 3,030 counties was only 0.60
percentage points, with a $t$ test on the full dataset yielding $t$(2422.6) = -1.50, $p$
= .133, though this result should be read with the caveat that nearly all
counties were represented in the sample. The value of Causal Mplus Trees
in analyzing these data is evident in its ability to find a group of counties
exhibiting stronger party effects, while simultaneously matching on COVID-19
trajectories.
Our findings corroborate those of previous COVID-19 partisanship studies.
Allcott et al. (2020) found evidence of 3.6 percent fewer point of interest visits
associated with a 10 percentage point decrease in the Republican vote share (roughly
equivalent to shifting from the median to the 25th percentile Republican vote share
county for the 2010 presidential election). Brzezinski et al. (2020) estimated a 3
percentage point difference in the share of devices staying fully at home for
the 90th vs 10th percentile Democrat vote share counties 15 days after a
county’s first case. Areas with relatively greater viewership of conservative
news shows that initially downplayed the threat of coronavirus (versus those
that accurately portrayed the pandemic) have also been linked to delayed
behavior changes and higher initial occurrences of cases and deaths (Bursztyn,
Rao, Roth, & Yanagizawa-Drott, 2020). Further, our Group 2 CATE is
comparable in magnitude to the decline in travel distance attributable to
statewide stay-at-home mandates (Sears et al., 2020). While prior studies
employ traditional approaches for discussing treatment effect heterogeneity (i.e.
running difference-in-differences or event study regressions on subgroups of
interest), the Causal Mplus Trees method provides a data-driven approach to
identifying comparable groups on model fit and analyzing treatment effect
heterogeneity.
In this paper, we proposed the Causal Mplus Trees algorithm, which matches on
parameter estimates of an SEM using a tree-based approach and uses these groupings
to estimate CATEs in a holdout sample. We used two small simulation studies to
demonstrate a proof of concept for the approach. We also showed how it could be
used to estimate party effects on mobility using COVID-19 data. We reiterate that we
do not see Causal Mplus Trees as a substitute for traditional matching methods.
Propensity score matching and related methods have their place and can be effective
in matching on covariates, both observed and latent. We believe that our
approach offers an alternative option to those whose research questions would be
better addressed by the ability to match on parameter estimates from an
SEM.
We encourage users of Causal Mplus Trees to carefully consider how they select and
differentiate between modeled covariates and splitting covariates. Although the
procedure ultimately matches on both, the way it does so differs by covariate
type. Matching is performed on modeled covariates indirectly through the
parameter estimates produced by the model, whereas splitting covariates
are matched more directly on the observed values of the scores. The choice
of whether a covariate should be used as a modeled or splitting covariate
depends upon what specifically the user wants to match, which can vary based
on the research question, study design, and characteristics of the sample
collected.
Another consideration for researchers using Causal Mplus Trees is the depth to
which the tree should be grown. Cross-validation is the most commonly used
approach for this in the context of conventional decision trees. However, we believe
that cross-validation may not be as well suited for our purposes primarily because it
is designed to optimize predictive accuracy. In our algorithm, the goal of the tree is
not to optimize predictive accuracy, but rather to partition the sample into groups
that are matched well enough on $\hat {\boldsymbol {\theta }}$ to justify causal inference in the holdout sample.
As in propensity score matching, there is no objective criterion for this, so
the researcher must make a subjective judgment and make a case to justify
it.
We urge researchers to take into account the following considerations. First, the
sample size in each parent node must be large enough to estimate $M$ in not only the
parent node, but also each of the daughter nodes. SEMs can require larger sample
sizes to estimate, so limits should be placed on the splitting procedure so as not to
consider splitting on a sample that does not have a large enough sample to do this.
Related to this is the need for a sufficient number of treated and untreated
participants in each terminal node to be able to estimate the CATEs in the
holdout sample. If a group has no treated (or no untreated) participants, the
CATE cannot be estimated. Of course, it is possible that the mix in the
tree differs from the mix in the holdout sample, but to the extent that the
matching subsample is a reflection of the estimation subsample, the matching
subsample can give a sense of the mix one would expect in the estimation
subsample. If performing hypothesis tests, certain minimum sample sizes are
required to meet the assumptions of the test as well as to detect the effects,
so these must also be kept in mind when deciding how deep to grow the
tree.
Parsimony is also important to consider, especially with respect to building a
coherent narrative with policy implications. We are typically searching for groups
with qualitative meaning given the relevant theoretical framework. If the tree
were to produce a dozen groups, it may be challenging to map this onto
available theory in order to interpret the results. The relative importance of
parameters in characterizing a pattern should be taken into account as well.
Theory may dictate that some parameters may be more important to match on
than others for a given context (e.g., the residual variance in our stability
example). As such, it could be justifiable to trim the tree earlier if splits begin
resulting in differences in less relevant parameters. The size of parameter
estimates may also play a role. For example, the algorithm could decide
on a split that results in two daughter nodes with only small differences in
their parameter estimates. Treating these as two separate groups for the
purpose of estimating the CATE may not be worthwhile. Similar to the logic
used in propensity score analysis, the treated and untreated participants in
each node should be compared on their parameters estimates, to verify, even
if only subjectively, that they are similar and therefore matched to some
degree.
The choice for the depth of the tree depends on a trade-off between interpretability of
a result and the validity of the causal inference. If one were to view the ability to
draw causal inference as how well treated and untreated participants are matched,
then the ability to draw causal inference can be conceptualized not as a dichotomy
but as a continuum with perfectly matched participants on one end and perfectly
unmatched participants on the other. The better matched participants are,
the greater the ability to draw causal inference. However, better matching
requires a deeper tree, which becomes less interpretable and generalizable as the
depth grows. This trade-off exists in propensity score matching as well but is
more apparent in the context of decision trees where such trade-offs are more
apparent and a language with which to conceptualize and discuss them already
exists.
Plenty of opportunities exist to expand on this work. Although two simulation
studies were conducted, they only served as a proof of concept. Additional
simulations would be helpful in evaluating the effectiveness of the algorithm
across a variety of conditions. The causal tree approach has been extended to
use random forests (Wager & Athey, 2018), which are known to be more
stable than decision trees. These causal forests have also been modified to
accommodate multilevel data structures (Suk, Kang, & Kim, in press). SEM Trees
have been expanded to SEM Forests (Brandmaier, Prindle, McArdle, &
Lindenberger, 2016), so expanding our algorithm to use random forests would be a
natural next step. Additionally, we note that our discussion of treatment effects
was limited to mean differences in univariate outcomes. However, given that
SEM is already being employed as well as the flexibility of Causal Mplus
Trees, it is possible that the outcome measure could be generalized to the
multivariate context, with treated and untreated participants being compared on a
model using, for example, a multiple group SEM. To conclude, we believe our
proposed algorithm can provide researchers with the opportunity to match on
SEM parameter estimates, thereby allowing them greater flexibility in what
they can match on and the kinds of research questions they can address as a
result.
Abrevaya, J., Hsu, Y.-C., & Lieli, R.
(2015). Estimating conditional average treatment effects. Journal of Business and
Economic Statistics, 33, 485–505. doi: https://doi.org/10.1080/07350015.2014.975555
Adolph, C., Amano, K., Bang-Jensen, B., Fullman, N., & Wilkerson,
J.
(2020). Pandemic politics: Timing state-level social distancing responses to
COVID-19. medRxiv. doi: https://doi.org/10.1101/2020.03.30.20046326
Allcott, H., Boxell, L., Conway, J., Gentzkow, M., Thaler, M., & Yang,
D.
(2020). Polarization and public health: Partisan differences in social distancing
during the coronavirus pandemic. Journal of Public Economics, 191. doi:
https://doi.org/10.1016/j.jpubeco.2020.104254
Athey, S., & Imbens, G.
(2016). Recursive partitioning for heterogeneous causal effects.
Proceedings of the National Academy of Sciences, 113, 7353–7360. doi:
https://doi.org/10.1073/pnas.1510489113
Austin, P.
(2009). The relative ability of different propensity-score methods to
balance measured covariates between treated and untreated subjects in
observational studies. Medical Decision Making, 29, 661–677. doi:
https://doi.org/10.1177/0272989X09341755
Austin, P.
(2011). An introduction to propensity score methods for reducing the
effects of confounding in observational studies. Multivariate Behavioral
Research, 46, 399–424. doi: https://doi.org/10.1080/00273171.2011.568786
Berk, R.
(2004). Regression analysis: A constructive critique. Sage. doi:
https://doi.org/10.4135/9781483348834
Brandmaier, A., Oertzen, T., McArdle, J., & Lindenberger, U.
(2013). Structural equation model trees. Psychological Methods, 18, 71–86.
doi: https://doi.org/10.1037/a0030001.
Brandmaier, A., Prindle, J., & Arnold, M.
(2021). semtree: Recursive partitioning for structural equation models [R package
version 0.9.17.]. Retrieved from https://CRAN.R-project.org/package=semtree
Brandmaier, A., Prindle, J., McArdle, J., & Lindenberger, U.
(2016). Theory-guided exploration with structural equation model forests.
Psychological Methods, 21, 566–582. doi: https://doi.org/10.1037/met0000090
Breiman, L., Friedman, J., Olshen, R., & Stone, C.
(1984). Classification and regression trees. Chapman & Hall. doi:
https://doi.org/10.1201/9781315139470
Browne, M., & Toit, S.
(1991). Models for learning data. In L. Collins & J. Horn (Eds.), Best
methods for the analysis of change (p. 47–68). American Psychological
Association. doi: https://doi.org/10.1037/10099-004
Brzezinski, A., Deiana, G., Kecht, V., & Van Dijcke, D.
(2020). The covid-19 pandemic: Government vs. community action
across the united states. INET Oxford Working Paper. Retrieved
from https://www.inet.ox.ac.uk/publications/no-2020-06-the-covid-19-pandemic-government-vs-community-action-across-the-united-states/
(No. 2020-06.)
Bursztyn, L., Rao, A., Roth, C., & Yanagizawa-Drott, D.
(2020). Misinformation during a pandemic. NBER Working Paper(27417).
doi: https://doi.org/10.3386/w27417
Dong, E., Du, H., & Gardner, L.
(2020). An interactive web-based dashboard to track covid-19 in real time. Lancet
Infectious Disease, 20, 533–534. doi: https://doi.org/10.1016/S1473-3099(20)30120-1
Ferrer, E.
(2016). Exploratory approaches for studying social interactions, dynamics, and
multivariate processes in psychological science. Multivariate Behavioral
Research, 51, 240–256. doi: https://doi.org/10.1080/00273171.2016.1140629
Ferrer, E., Steele, J., & Hsieh, F.
(2012). Analyzing dynamics of affective dyadic interactions using
patterns of intra- and inter-individual variability. Multivariate Behavioral
Research, 47, 136–171. doi: https://doi.org/10.1080/00273171.2012.640605
Gadarian, S., Goodman, S., & Pepinsky, T.
(2020). Partisanship, health behavior, and policy attitudes in the early stages
of the COVID-19 pandemic. SSRN. doi: https://doi.org/10.2139/ssrn.3562796
Greenland, S., Pearl, J., & Robins, J.
(1999). Causal diagrams for epidemiologic research. Epidemiology,
10, 37–48. doi: https://doi.org/10.1097/00001648-199901000-00008
Grimm, K., & Ram, N.
(2009). Nonlinear growth models in mplus and sas. Structural Equation
Modeling, 16, 676–701. doi: https://doi.org/10.1080/10705510903206055
Gu, X., & Rosenbaum, P.
(1993). Comparison of multivariate matching methods: Structures,
distances, and algorithms. Journal of Computational and Graphical
Statistics, 2, 405–420. doi: https://doi.org/10.1080/10618600.1993.10474623
Guo, S., & Fraser, M.
(2010). Propensity score analysis: Statistical methods and applications.
Sage.
Hallquist, M., & Wiley, J.
(2018). MplusAutomation: An R package for facilitating large-scale latent
variable analyses in Mplus. Structural Equation Modeling, 25, 621–638. doi:
https://doi.org/10.1080/10705511.2017.1402334
Harder, V., Stuart, E., & Anthony, J.
(2010). Propensity score techniques and the assessment of measured
covariate balance to test causal associations in psychological research.
Psychological Methods, 15, 234–249. doi: https://doi.org/10.1037/a0019623
Hirano, K., & Imbens, G.
(2001). Estimation of causal effects using propensity score weighting: An
application to data on right heart catheterization. Health Services and Outcomes
Research Methodology, 2, 259–278. doi: https://doi.org/10.1023/A:1020371312283
Ho, D., Imai, K., King, G., & Stuart, E.
(2007). Matching as nonparametric preprocessing for reducing model
dependence in parametric causal inference. Political Analysis, 15, 199–236. doi:
https://doi.org/10.1093/pan/mpl013
Holland, P.
(1986). Statistics and causal inference. Journal of the American Statistical
Association, 81, 945–60. doi: https://doi.org/10.1080/01621459.1986.10478354
Jöreskog, K.
(1971). Simultaneous factor analysis in several populations. Psychometrika,
36, 409-426. doi: https://doi.org/10.1007/BF02291366
Lee, B., Lessler, J., & Stuart, E.
(2010). Improving propensity score weighting using machine learning.
Statistics in Medicine, 29, 337–346. doi: https://doi.org/10.1002/sim.3782
Leite, W., Stapleton, L., & Bettini, E.
(2018). Propensity score analysis of complex survey data with structural
equation modeling: A tutorial with mplus. Structural Equation Modeling, 3,
448–469. doi: https://doi.org/10.1080/10705511.2018.1522591
Meredith, W., & Tisak, J.
(1990). Latent curve analysis. Psychometrika, 55, 107–122. doi:
https://doi.org/10.1007/bf02294746
Muthén, L., & Muthén, B.
(1998-2017). Mplus user’s guide (8th ed.) [Computer software manual].
Muthén & Muthén.
National Governors Association.
(2020). Governors roster. Retrieved
from https://www.nga.org/wp-content/uploads/2019/07/Governors-Roster.pdf
Neale, M., Hunter, M., Pritikin, J., Zahery, M., Brick, T., Kirkpatrick, R., &
Boker, S.
(2016). Openmx 2.0: Extended structural equation and statistical modeling.
Psychometrika, 81, 535–549. doi: https://doi.org/10.1007/s11336-014-9435-8
Neyman, J., Iwaszkiewicz, K., & Kolodziejczyk, S.
(1935). Statistical problems in agricultural experimentation. Supplement
to the Journal of the Royal Statistical Society, 2, 107–180. doi:
https://doi.org/10.2307/2983637
Normand, S., Landrum, M., Guadagnoli, E., Ayanian, J., Ryan, T., Cleary, P., &
McNeil, B.
(2001). Validating recommendations for coronary angiography following an
acute myocardial infarction in the elderly: A matched analysis using
propensity scores. Journal of Clinical Epidemiology, 54, 387–398. doi:
https://doi.org/10.1016/S0895-4356(00)00321-8
R Core Team.
(2020). R: A language and environment for statistical computing [Computer
software manual]. Vienna, Austria. Retrieved from https://www.R-project.org/
Raykov, T.
(2012). Propensity score analysis with fallible covariates: A note on
a latent variable modeling approach. Educational and Psychological
Measurement, 72, 715–733. doi: https://doi.org/10.1177/0013164412440999
Rosenbaum, P., & Rubin, D.
(1983). The central role of the propensity score in observational studies for causal
effects. Biometrika, 70, 41–55. doi: https://doi.org/10.1093/biomet/70.1.41
Rosenbaum, P., & Rubin, D.
(1984). Reducing bias in observational studies using subclassification on the
propensity score. Journal of the American Statistical Association, 79, 516–24.
doi: https://doi.org/10.2307/2288398
Rosenbaum, P., & Rubin, D.
(1985). The bias due to incomplete matching. Biometrics, 41, 103–16. doi:
https://doi.org/10.2307/2530647
Rosseel, Y.
(2012). lavaan: An R package for structural equation modeling. Journal of
Statistical Software, 48(2), 1–36. doi: https://doi.org/10.18637/jss.v048.i02
Rubin, D.
(1974). Estimating causal effects of treatments in randomized and
nonrandomized studies. Journal of Educational Psychology, 66, 688–701. doi:
https://doi.org/10.1037/h0037350
Rubin, D.
(1980). Randomization analysis of experimental data: The fisher
randomization test comment. Journal of the American Statistical Association,
75, 591–593. doi: https://doi.org/10.2307/2287653
Rubin, D.
(1986). What if’s have causal answers. Journal of the American Statistical
Association, 81, 961–962. doi: https://doi.org/10.1080/01621459.1986.10478355
Sears, J., Villas-Boas, S., Villas-Boas, M., & Villas-Boas, V.
(2020). Are we #stayinghome to flatten the curve? SSRN. doi:
https://doi.org/10.2139/ssrn.3569791
Serang, S., Jacobucci, R., Stegmann, G., Brandmaier, A., Culianos, D., & Grimm,
K.
(2021). Mplus Trees: Structural equation model trees using Mplus. Structural
Equation Modeling, 28, 127–137. doi: https://doi.org/10.1080/10705511.2020.1726179
Stegmann, G., Jacobucci, R., Serang, S., & Grimm, K.
(2018). Recursive partitioning with nonlinear models of change. Multivariate
Behavioral Research, 53, 559–570. doi: https://doi.org/10.1080/00273171.2018.1461602
Suk, Y., Kang, H., & Kim, J.-S.
(in press). Random forests approach for causal inference with
clustered observational data. Multivariate Behavioral Research. doi:
https://doi.org/10.1080/00273171.2020.1808437
Therneau, T., & Atkinson, B.
(2018). Rpart: Recursive partitioning and regression trees [R package version
4.1-13.]. Retrieved from https://CRAN.R-project.org/package=rpart
Thoemmes, F., & Kim, E.
(2011). A systematic review of propensity score methods in the
social sciences. Multivariate Behavioral Research, 46, 90–118. doi:
https://doi.org/10.1080/00273171.2011.540475
Unacast.
(2020). Unacast social distancing scoreboard dataset. Retrieved
from https://www.unacast.com/data-for-good.
U.S. Census Bureau.
(2010). Decennial census, 2010. Retrieved from https://data.census.gov/
U.S. Department of Education, Institute of Education Sciences, & What Works
Clearinghouse.
(2017). What works clearinghouse: Standards handbook (version 4.0). Retrieved
from https://ies.ed.gov/ncee/wwc/Docs/referenceresources/wwc\_standards\_\\handbook\_v4.pdf
Wager, S., & Athey, S.
(2018). Estimation and inference of heterogeneous treatment effects
using random forests. Journal of the American Statistical Association,
113, 1228–1242. doi: https://doi.org/10.1080/01621459.2017.1319839
West, S., Cham, H., Thoemmes, F., Renneberg, B., Schulze, J., & Weiler,
M.
(2014). Propensity scores as a basis for equating groups: Basic principles and
application in clinical treatment outcome research. Journal of Consulting and
Clinical Psychology, 82, 906–919. doi: https://doi.org/10.1037/a0036387
1.2 Purpose
1.3 Propensity Score Matching
1.4 Causal Trees
1.5 Structural Equation Model Trees
1.6 Mplus Trees
2 Causal Mplus Trees
3 Simulation Studies
3.1 Longitudinal Simulation
3.2 Measurement Simulation
4 Empirical Example

(2)
Table 1: Parameter Estimates for SEMs from the Groups in Figure 4
Group 1 Group 2 Group 3 Group 4
$n$ 696 362 744 622
$\beta _1$ (Mean) 61.33 50.19 35.88 19.04
$\beta _1$ (Variance) 13,377.34 7,767.81 2,425.19 455.58
$\gamma $ 9.33 6.94 10.23 10.44
$\alpha $ 0.69 0.56 0.44 0.40
$\sigma _\epsilon ^2$ 554.05 139.86 88.02 18.73
Table 2: CATEs and Significance Tests for COVID-19 Groups
Group 1 Group 2 Group 3 Group 4
$n_{Rep} ; n_{Dem} $ 104; 59 58; 55 104; 94 76; 60
$\overline {CADT}_{Rep}$ -0.93% -4.47% -0.58% -0.78%
$\overline {CADT}_{Dem}$ -2.47% -10.92% 0.88% -2.30%
CATE 1.54% 6.46% -1.46% 1.53%
$t$ test $t$(104.08) = 0.94 $t$(108.76) = 2.84 $t$(186.87) = -0.86 $t$(122.99) = 1.15
$p$ value .349 .006 .389 .252
5 Discussion
5.1 Practical Recommendations
5.2 Future Research and Conclusions
References